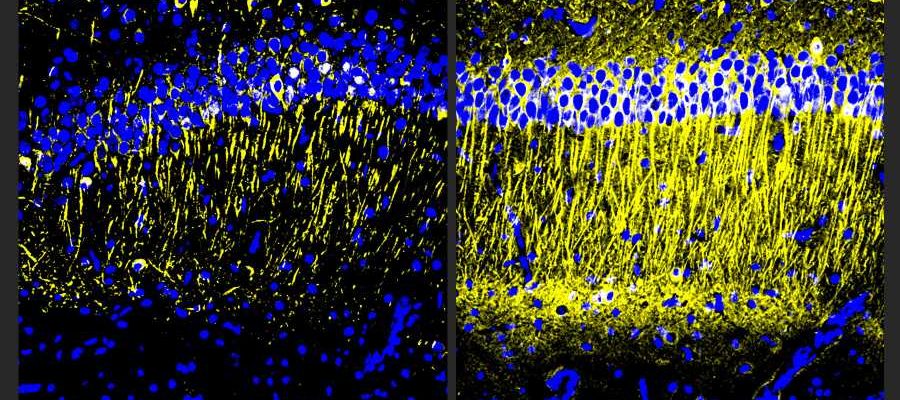
 showed more tubulin (yellow), a marker of neuronal health, than untreated controls (left). Credit: Tsai Lab/MIT Picower Institute")
Though drug developers have achieved some progress in treating Alzheimer’s disease with medicines that reduce amyloid-beta protein, other problems of the disease including inflammation, continue unchecked. In a new study, scientists at The Picower Institute for Learning and Memory at MIT describe a candidate drug that in human cell cultures and Alzheimer’s mouse models reduced inflammation and improved memory.
The target of the new “A11” molecule is a genetic transcription factor called PU.1. Prior research has shown that amid Alzheimer’s disease, PU.1 becomes an overzealous director of inflammatory gene expression in the brain’s microglia immune cells. A11 suppresses this problematic PU.1 activity, the new research shows, by recruiting other proteins that repress the inflammatory genes PU.1 works to express.
But because A11 concentrates mostly in the brain and does not reduce PU.1 levels, it does not appear to disrupt PU.1’s other job, which is to ensure the production of a wide variety of blood cells.
“Inflammation is a major component of Alzheimer’s disease pathology that has been especially hard to treat,” said study senior author Li-Huei Tsai, Picower Professor of Neuroscience at MIT and director of The Picower Institute and MIT’s Aging Brain Initiative. “This preclinical study demonstrates that A11 reduces inflammation in human microglia-like cells as well as in multiple mouse models of Alzheimer’s disease and significantly improves cognition in the mice. We believe A11 therefore merits further development and testing.”
Tsai and Elizabeta Gjoneska of the National Institutes of Health are co-corresponding authors of the study published in the Journal of Experimental Medicine.
As a postdoc, Gjoneska co-led a 2015 study that implicated PU.1 as a regulator of errant microglia inflammation in a mouse model of Alzheimer’s disease. That research was a collaboration between Tsai’s lab and that of MIT Computer Science Professor Manolis Kellis, co-led by former postdoc Andreas Pfenning, now a faculty member at Carnegie Mellon University. Ever since then, Tsai has been seeking a safe way to restore PU.1 activity to healthier levels.
The work described in the new paper, led by Picower Institute research scientist William Ralvenius, starts with experiments to further validate that PU.1 would be a therapeutically meaningful target. To do that the scientists compared gene expression in immune cells of postmortem brain samples from Alzheimer’s patients and mouse models and matching non-Alzheimer’s controls.
The comparisons showed that Alzheimer’s effects major changes in microglial gene expression and that an increase in PU.1 binding to inflammatory gene targets was a significant component of that change. Moreover, they showed that reducing PU.1 activity in a mouse model of Alzheimer’s reduced inflammation and neurodegeneration, the death of neurons.
Screening success
Genetically knocking down PU.1 in the body is not a viable therapeutic strategy given its importance in normal healthy function. The team therefore screened more than 58,000 small molecules from libraries of FDA-approved drugs and novel chemicals to see if any could safely and significantly reduce key inflammation and Alzheimer’s related genes regulated by by PU.1 in cell cultures. After several rounds of increasingly stringent screening, they narrowed the field down to six chemicals. A11 was by far the most potent among them.
They tested the effects of A11 doses on the function of human microglia-like cells cultured from patient stem cells. When they exposed the microglia-like cells to immune molecules that typically trigger inflammation, cells dosed with A11 exhibited reduced expression and secretion of inflammatory cytokines and less of the cell body shape changes associated with microglia inflammatory responses.
The cells also showed less accumulation of lipid molecules, another sign of inflammatory activation. Looking at gene expression patterns, the scientists observed that A11-treated cells exposed to inflammatory triggers behaved much like unperturbed microglia, suggesting that A11 helps prevent microglia from overreacting to inflammatory cues.
Two more lab tests aimed at understanding how A11 exerts its effects revealed that it doesn’t change PU.1 levels. Instead it counteracts PU.1 activity by recruiting several proteins including MECP2, HDAC1, SIN3A and DMNT3A, known to repress expression of its targets. Essentially amid Alzheimer’s disease, A11 tamps down what PU.1 amps up.
“A11 represents a first-in-class molecule that converts PU.1 from a transcriptional activator to a transcriptional repressor, resulting in a controlled state of microglial inflammation,” the authors wrote.
Mice in mazes
Having established that A11 reduced inflammatory activity in microglia and determined how that happens, the team focused on whether it worked as a medicine in mouse models of Alzheimer’s disease.
Pharmacological tests indicated that A11 is readily cleared from tissues and is capable of reaching brain cells. Moreover, in healthy mice the chemical successfully crossed the blood-brain barrier and remained in brain cells much longer than anywhere else.
Finally the team tested the effects of the drugs on Alzheimer’s disease pathology and symptoms in three mouse strains that each model different aspects of Alzheimer’s disease: CK-p25 mice (severe neurodegeneration), Tau P301S transgenic mice (tauopathy), and 5XFAD mice (amyloid pathology).
Male and female CK-p25 mice dosed with A11 showed less inflammatory response among microglia and astrocyte cells and lost fewer neurons than untreated controls. TauP301S Tg mice responded similarly, also exhibiting a significant reduction of phosphorylated tau protein in the hippocampus region of the brain, which is an essential area for memory. In 5XFAD mice, amyloid was significantly reduced.
The team subjected the Tau P301S Tg and CK-p25 mice to mazes designed to test their short-term working memory and longer-term learning. In both models and on both tests, A11-treated mice performed significantly better than untreated controls. For example, in the “Morris Water Maze,” where mice have to learn the location of a submerged platform that allows them to rest, treated CK-p25 mice learned much faster than untreated ones.
Much more testing needs to be done before A11 could become an approved medicine, Tsai said, but she noted that it could complement the new treatments that target amyloid.
“Given that A11 acts via a distinct mechanism from existing AD therapeutics, A11 could be used alone or in combination with approved therapeutics to provide improved treatment options for neurodegenerative diseases,” the authors concluded.
More information:
William Ralvenius et al, A novel molecular class that recruits HDAC/MECP2 complexes to PU.1 motifs reduces neuroinflammation, Journal of Experimental Medicine (2023). DOI: 10.1084/jem.20222105
Journal information:
Journal of Experimental Medicine
Source: Read Full Article